Расширенная российскими учеными методика секвенирования поможет делать качественные «геномные портреты» бактерий
Российские ученые оценили состав кишечного сообщества бактерий, которые при ослаблении иммунитета могут вызывать инфекции у пациентов, в течение длительного времени пребывающих в реанимации. Для получения качественного «геномного портрета» бактерий они применили технологию Hi-C метагеномики, представляющую собой сочетание обычного метагеномного ДНК-секвенирования с технологией определения «трехмерного генома».
Проведенное исследование — одно из первых в мире с клиническим применением Hi-C метагеномики. В перспективе с его помощью ученые смогут больше узнавать о составе и функциях микробиома людей. На основании анализа большой коллекции геномов станет возможным создание более дешевых и быстрых методов диагностики инфекций. Применять их можно будет непосредственно в больнице, что крайне важно для сохранения жизни людей в реанимации.
В исследовании приняли участие ученые из Института биологии гена (ИБГ) РАН, Московского государственного университета им. М.В. Ломоносова, Центра высокоточного редактирования и генетических технологий для биомедицины ИБГ РАН, Федерального научно-клинического центра реаниматологии и реабилитологии, Центра алгоритмической биотехнологии Санкт-Петербургского государственного университета, Университета ИТМО и Медико-генетического научного центра им. академика Н.П. Бочкова.
Микробное сообщество (микробиом) кишечника человека участвует в регуляции работы самых разных систем: пищеварительной, иммунной, гормональной, нервной и других. Это обеспечивается слаженным взаимодействием многих сотен видов микроорганизмов. Когда человек заболевает, баланс микробиома нарушается, что может вести к дальнейшему ухудшению состояния. Как отмечают ученые, исследование таких нарушений нужно для понимания механизмов возникновения и развития сахарного диабета, сердечно-сосудистых заболеваний, а также осложнений при COVID-19. В перспективе на основе этих наблюдений можно будет высокоточно управлять составом микробиома, контролируя его и таким образом снижая риск заболеваний.
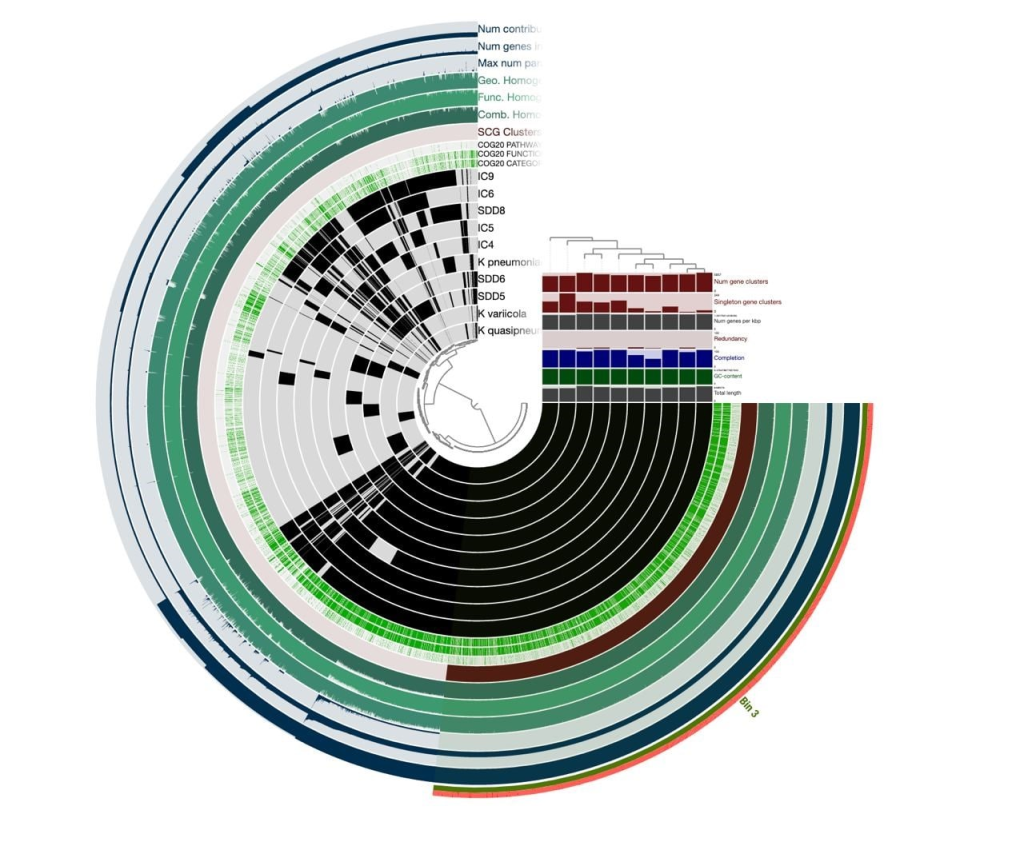
Для накопления наблюдательной базы по микробиому ученые используют метагеномику — технологию на основе ДНК-секвенирования микробиома. Это позволяет широко описать набор видов бактерий в каждом отдельном образце микробиома, при этом оценить долю каждого из них и выяснить его функции. Однако из метагеномных данных не всегда удается получить качественный «геномный портрет» той или иной бактерии. Поэтому был создан новый метод — Hi-C метагеномика. В его основе лежит метагеномное ДНК-секвенирование и технология определения пространственной формы генома (Hi-C; также известная как «3D-геномика»).
Российские ученые наладили комплексную экспериментально-биоинформатическую процедуру для Hi-C метагеномики и с ее помощью изучили самые разнообразные ниши, от сообществ горячих источников до пива спонтанного брожения.
«Одним из направлений исследований стал анализ микробиома больных в критических состояниях. У пациентов, длительно пребывающих в реанимации, наблюдаются серьезные нарушения состава микробиома. Сообщество бактерий, населяющее их кишечник, потенциально может содержать виды, которые при ослаблении иммунитета могут вызвать инфекции и повысить риск смерти. Поэтому важно изучать, какие виды есть у пациентов и какие особые гены они несут, например, дающие устойчивость к антибиотикам или повышающие их вирулентность (способность патогена или микроорганизма причинять вред хозяину)», — рассказал Александр Тяхт, руководитель проекта, кандидат биологических наук, заведующий группой биоинформатики ИБГ РАН.
Hi-C метагеномика позволяет исследователям получать более полные и точные геномы бактерий. С помощью графовых алгоритмов ученым удалось более точно отнести к той или иной бактерии мобильные генетические элементы — фрагменты ДНК, которые часто передаются между разными видами бактерий и способствуют распространению генов лекарственной устойчивости и других биомедицински важных генов между бактериями — возбудителями заболеваний.
Дальнейшее применение метода позволит не только подробнее узнать геномы возбудителей инфекций, но также лучше связать мобильные генетические элементы с бактериями, которым они принадлежат. Подобная база знаний даст возможность разрабатывать прицельные методы для выявления опасных бактерий на базе, например, таких подходов, как ПЦР в реальном времени. Между тем, ученые уже применяют метод для анализа и других микробиомов — как пациентов с различными заболеваниями, так и из других ниш, например, окружающей среды и пищевых продуктов.
Научная статья опубликована в журнале Frontiers in Microbioligy. Исследование поддержано Российским научным фондом и Минобрнауки России.
Иллюстрация - Сравнение геномов бактерий — потенциальных возбудителей инфекций, выделенных из Hi-C метагенома.